This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison. https://genetics564.weebly.com/
Introduction
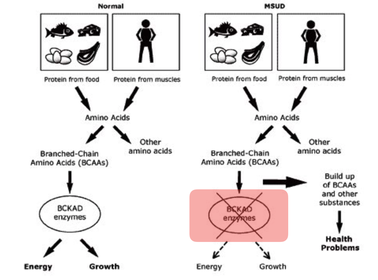
Maple Syrup Urine Disease (MSUD) is a genetic metabolic disorder that every individual in the United States is tested for in Newborn Screening tests. It results from the body’s inability to break down three amino acids: valine, leucine, and isoleucine. Initial symptoms lead to the sweet smell of maple syrup in urine, sweat, and ear wax. [1,2] MSUD is more common in populations with history of homozygosity, such as the Amish. [3]
One gene associated with Maple Syrup Urine Disease is DBT. DBT encodes for the E2 subunit of the branched chain alpha-ketoacid dehydrogenase, which localizes in the mitochondria. This is its cellular function. [1,4] The E2 subunit has been characterized as a core protein necessary for catalyzing the second step of the amino acid breakdown or acyltransferase step, and this is its molecular function. [5] Biologically, this is essential for the break down of the BCAA for the body to use in other metabolic cycles. [6]
Without treatment for prolonged time, the build up of the three amino acids can cause poor feeding, brain damage, developmental delays, seizure-like spasms, and can even be fatal. [1,2] The poor feeding symptom will be the focus of the research.
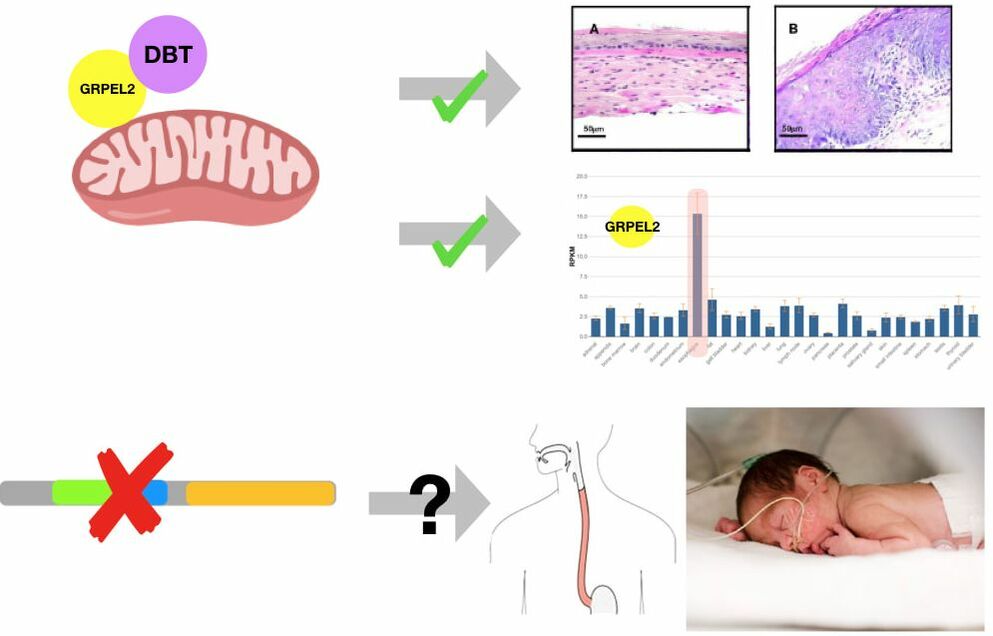
DBT is highly conserved across species. It contains three domains - the biotin lipoyl domain, the E3 domain, and the catalytic domain. Phylogenetic tree analysis of the DBT protein shows a distinct divergence of complex and simple digestive system organisms. Finally, protein interactions find something of great intrigue - there is a protein interaction between DBT and GRPEL2, which is highly expressed in the esophagus. [7] GRPEL2 is a mitochondrial membrane transport protein. [8] This led to the focus on the esophageal function.
One gene associated with Maple Syrup Urine Disease is DBT. DBT encodes for the E2 subunit of the branched chain alpha-ketoacid dehydrogenase, which localizes in the mitochondria. This is its cellular function. [1,4] The E2 subunit has been characterized as a core protein necessary for catalyzing the second step of the amino acid breakdown or acyltransferase step, and this is its molecular function. [5] Biologically, this is essential for the break down of the BCAA for the body to use in other metabolic cycles. [6]
Without treatment for prolonged time, the build up of the three amino acids can cause poor feeding, brain damage, developmental delays, seizure-like spasms, and can even be fatal. [1,2] The poor feeding symptom will be the focus of the research.
DBT is highly conserved across species. It contains three domains - the biotin lipoyl domain, the E3 domain, and the catalytic domain. Phylogenetic tree analysis of the DBT protein shows a distinct divergence of complex and simple digestive system organisms. Finally, protein interactions find something of great intrigue - there is a protein interaction between DBT and GRPEL2, which is highly expressed in the esophagus. [7] GRPEL2 is a mitochondrial membrane transport protein. [8] This led to the focus on the esophageal function.
Gap In Knowledge
|
It is known that DBT interacts with GRPEL2, which highly localizes in the mitochondria. It is also known that other disorders known to affect mitochondrial function, similar to MSUD, have lead to GERD, which will affect feeding and esophageal function. [9,10] However, although poor feeding is a known symptom, it is unclear how mutations in DBT lead to the poor feeding phenotype. Therefore, the primary goal of this study is determine how DBT plays a role in esophageal function in the early postnatal period. |
Specific Aim 1
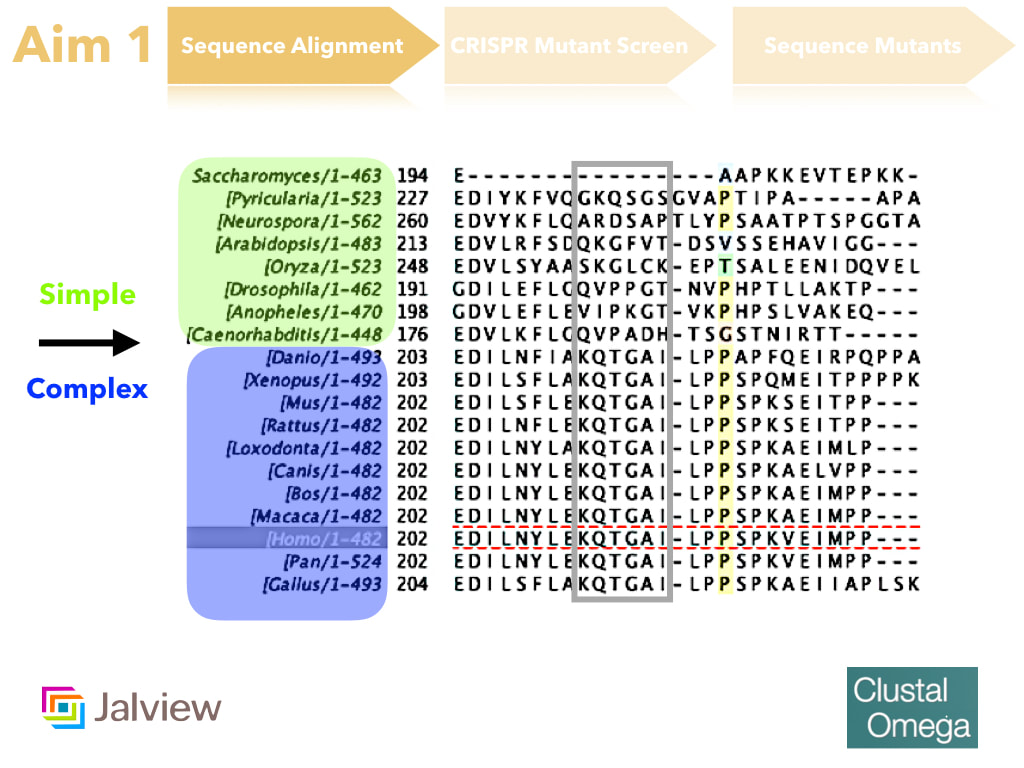
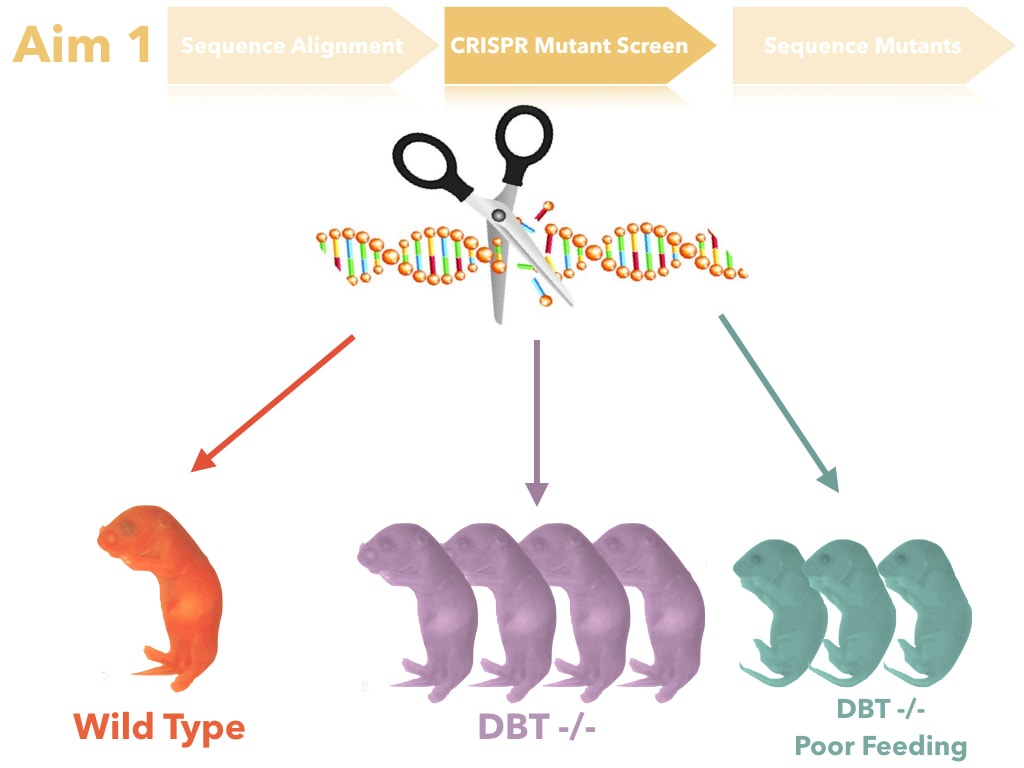
The goal of this aim is to identify the amino acids that are important for feeding. I hypothesize that conserved amino acids among complex digestive system organisms are important for feeding. I intend to determine this through sequence alignment and identification of amino acids that are conserved only in complex digestive species, but not simple. These amino acids will then be mutated through CRISPR-Cas9 mutation and assayed for esophageal tissue presentation, urine analysis, and feeding behavior (for more information on assay techniques, please see the Model Organisms Page). DBT mutants and DBT mutants with poor feeding will then undergo sequencing to verify that the correct amino acids were mutated and thus led to the poor feeding phenotype.
|
|
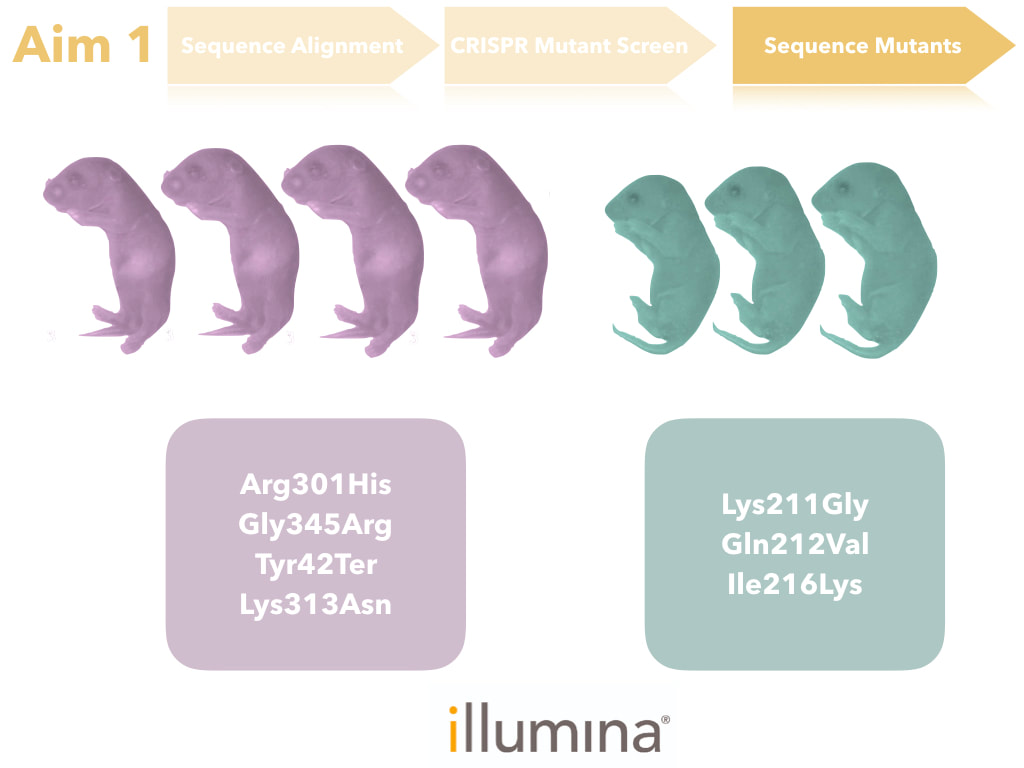
|
Specific Aim 2
The goal of this aim is to determine transcript levels that impact feeding. I hypothesize that the transcripts of genes in the mitochondria will have altered expression levels. In order to do this, transcript levels will be sequenced from esophageal tissues. Extracted tissues will undergo RNAsequence analysis. These will undergo expression profiling to form clusters of transcripts of similar gene ontology to see what groups of genes are up and down regulated. Genes of interest will be mutated in mice through CRISPR-Cas9 and assayed to determine if they will still produce the same phenotype.
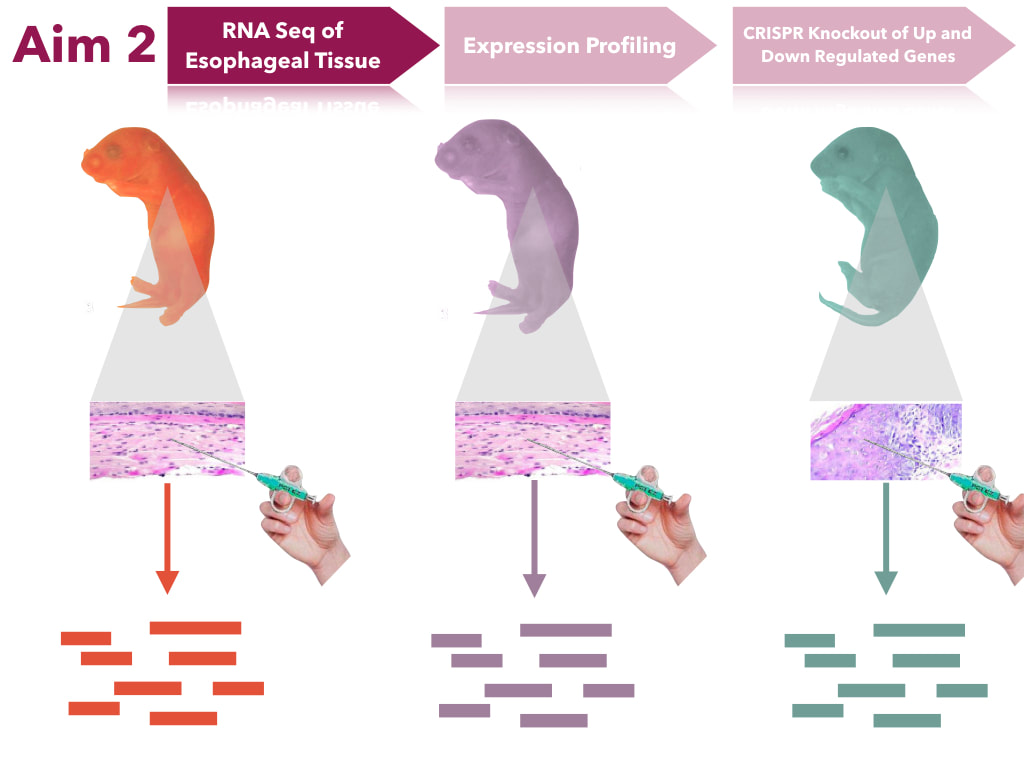
|
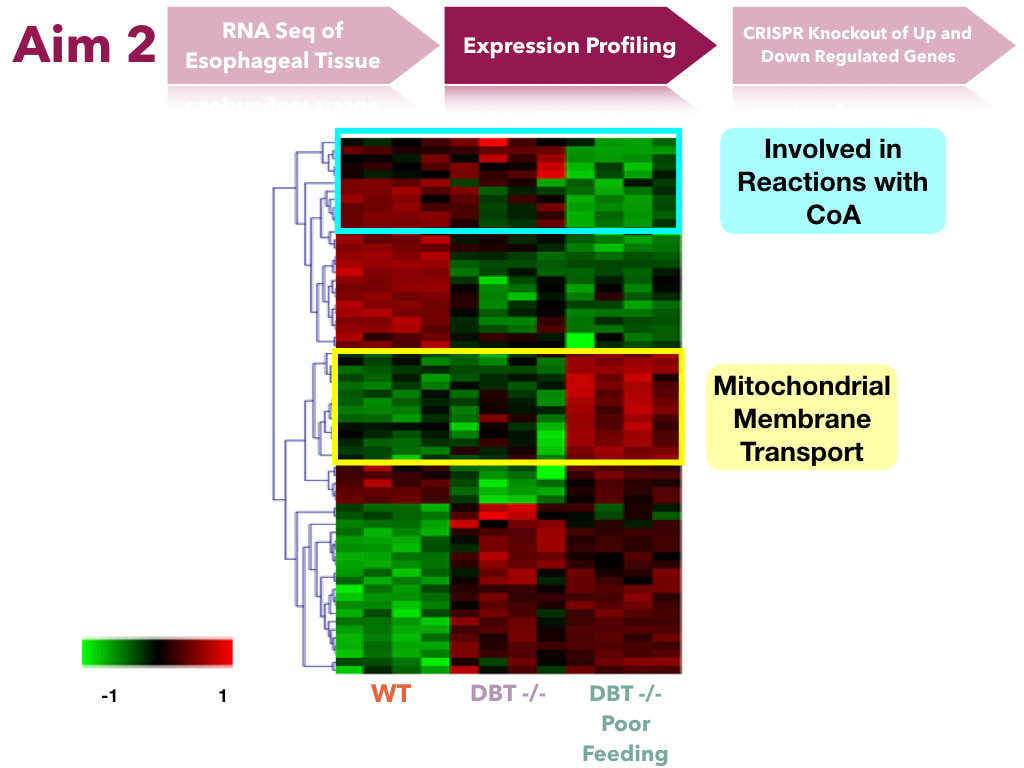
|
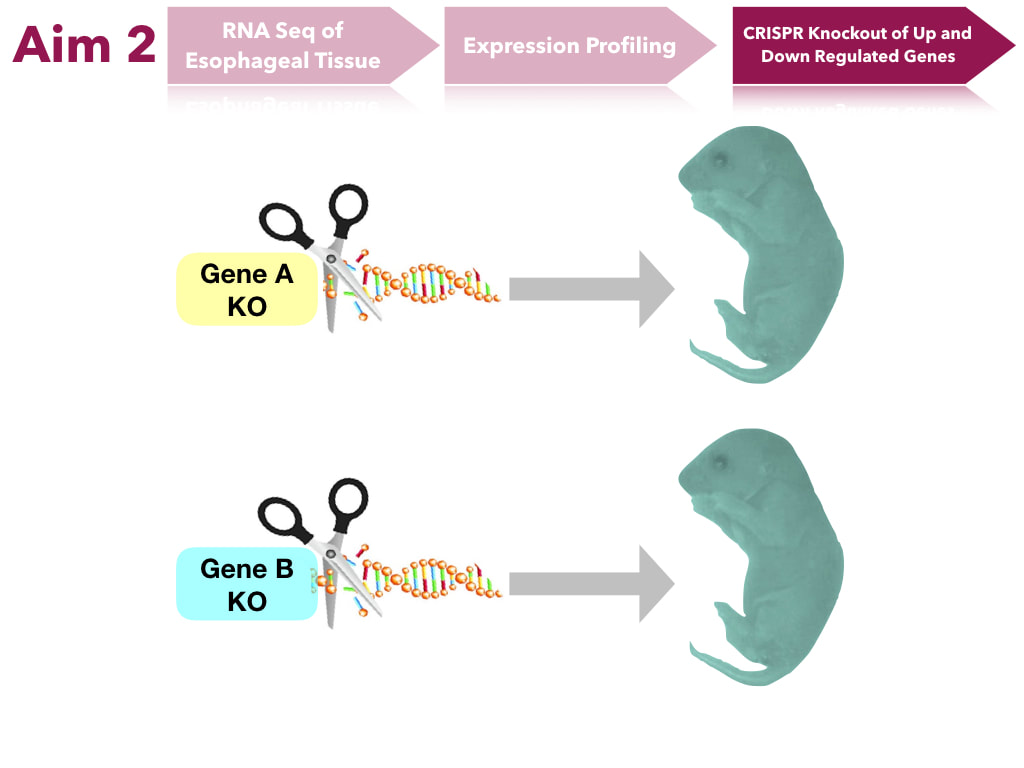
|
Specific Aim 3
The goal of this aim is to identify proteins necessary for proper feeding. I hypothesize that protein interactions in the mutant DBT with poor feeding will have loss of protein interaction with mitochondrial transport proteins. In order to do this, esophageal tissue will be extracted. Then, protein interactions with DBT will be determined through Tandem Affinity Purification, SDS Page, Trypsin Digestion, and then Mass Spectrometry. The protein interactions will then be mapped based on gene ontology. Proteins (or protein groups) of interest will be the ones that change interaction levels from both the Wild Type and the DBT mutants (no poor feeding). Those proteins will be mutated through CRISPR-Cas9 and those mice will assayed. If important for feeding, they will produce a poor feeding phenotype in the mutant mice.
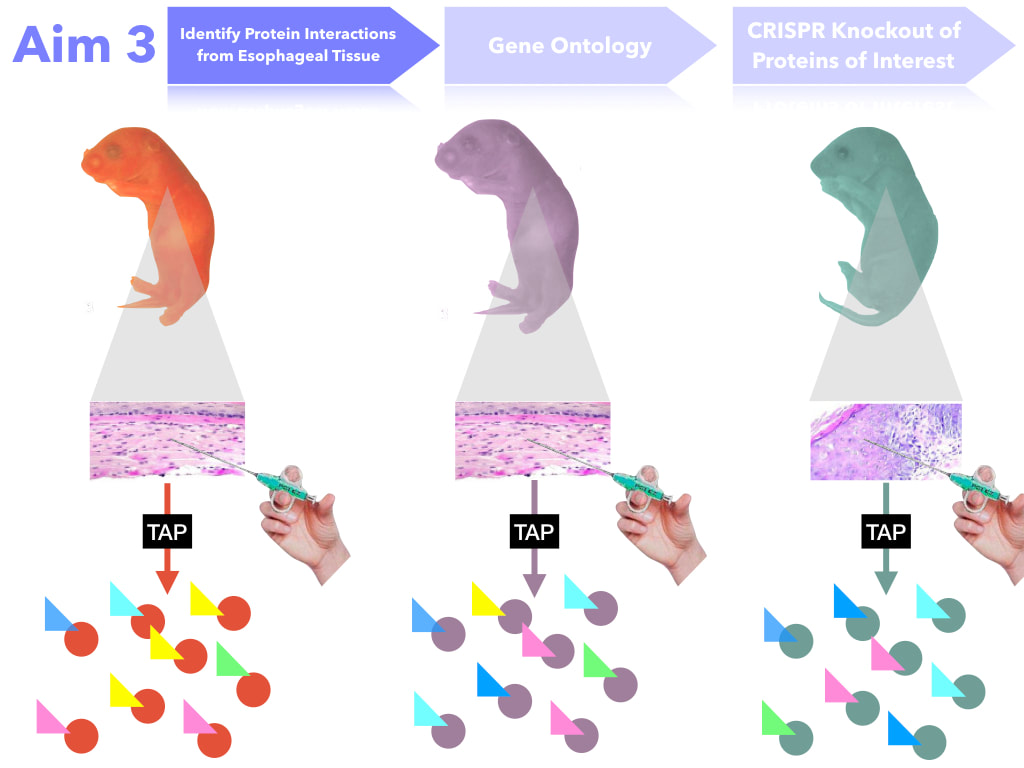
|
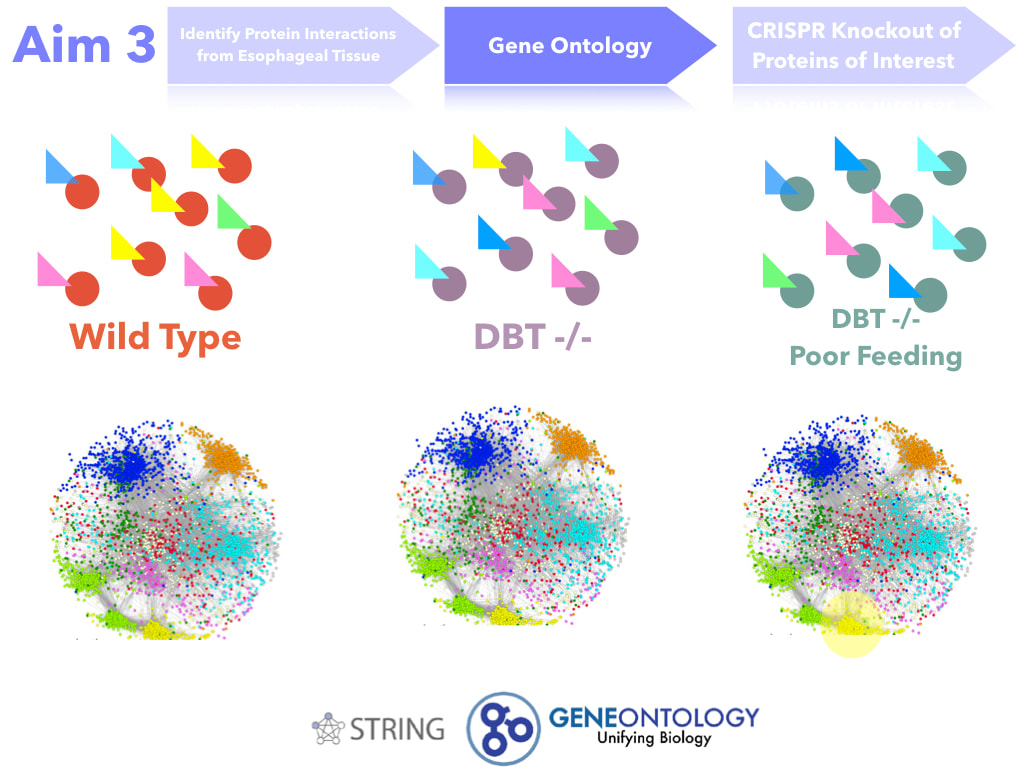
|
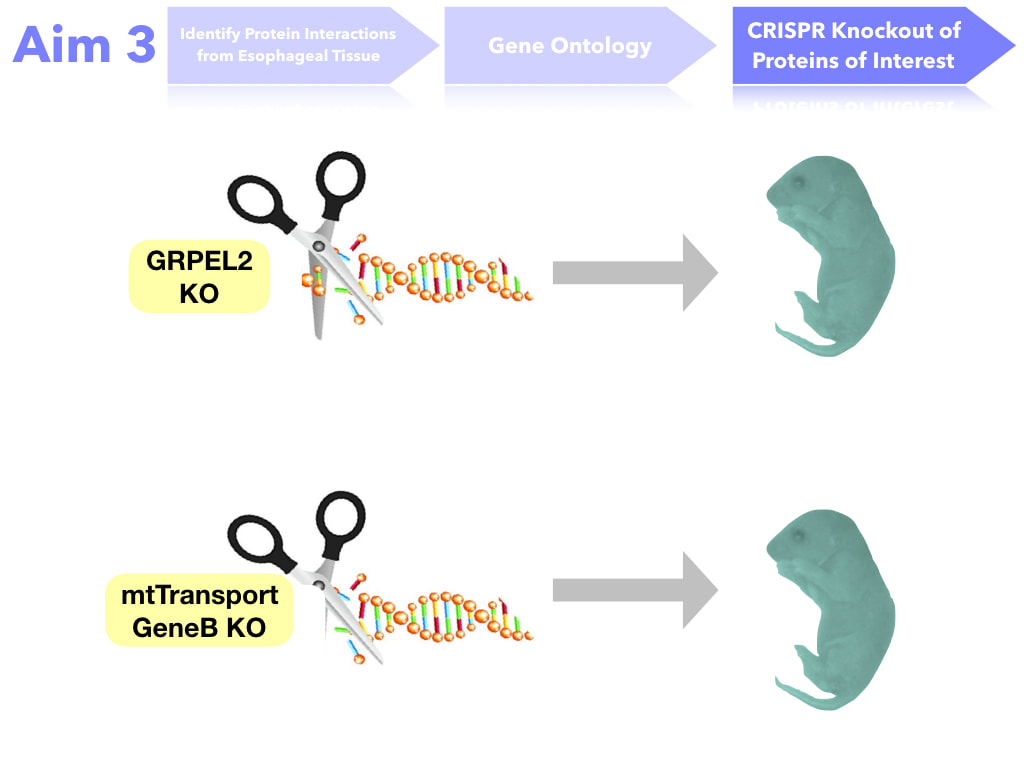
|
Conclusions
Mutations in the DBT gene leads to Maple Syrup Urine Disease. One symptom some children present with is poor feeding. Investigation of conserved amino acids, gene expression levels, and protein interactions can elucidate what pathogenic variants may be causing poor feeding in infants. In clinic, this can be used as important information in watching for poor feeding behavior and addressing it at an early stage.
Future Directions
Many amino acids conserved in complex digestive species were found to be in a region that is not part of a characterized domain, specifically the region from amino acid K211-I216. This region is between the E3 binding domain and the catalytic domain in the human protein. Investigating this further could characterize a new domain of the DBT protein, providing insight into pathogenic variants. Additionally, this information could be used in clinic to better address symptoms ahead of time based on where the patient’s variant is.
Drafts
Keynote Link: https://uwmadison.box.com/s/r2ompab0xqx0ookene0qm2xw4z4jd2ga
Powerpoint Link: https://uwmadison.box.com/s/8f8xodoa4ufog9h9wh6cl88gxn1ihauf
Powerpoint Link: https://uwmadison.box.com/s/8f8xodoa4ufog9h9wh6cl88gxn1ihauf
| bachinski_presentation_draft_1_4_5_19_3.pptx.zip |
| bachinski_presentation_draft_1_4_5_19_3.pdf |
Final Talk
| bachinski_final_talk_4_23_19_pdf.pdf |
References
[1] Strauss, K., Puffenberger, E., Morton, H. (May 2013). Maple Syrup Urine Disease. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1319/#msud.molgen.TA
[2] National Organization for Rare Disorders. (2017). Rare Disease Data Base: Maple Syrup Urine Disease. Retrieved from https://rarediseases.org/rare-diseases/maple-syrup-urine-disease/#supporting-organizations
[3] Abiri, M., Karamzadeh, R., Mojbafan, M., et al. (August 2016). In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Retrieved from https://link.springer.com/article/10.1007%2Fs11011-016-9867-1
[4] Human Protein Atlas. (n.d.) DBT. Retrieved from https://www.proteinatlas.org/ENSG00000137992-DBT/cell
[5] Evarsson, A., Chuang, J., Wynn, R., et al. (March 2000). Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Retrieved from https://www.sciencedirect.com/science/article/pii/S0969212600001052
[6] Kegg Pathway. https://www.genome.jp/kegg-bin/show_pathway?ko00280+K09699
[7] GRPEL2 GrpE like 2, mitochondrial [ Homo sapiens (human) ]. Retrieved from: https://www.ncbi.nlm.nih.gov/gene
[8] String. https://string-db.org/cgi/network.pl?taskId=IdKnOD6d1YVA
[9] Gilles, Hepple, & T., R. (2016, September 20). Editorial: Mitochondria in Skeletal Muscle Health, Aging and Diseases. Retrieved from https://www.frontiersin.org/articles/10.3389/fphys.2016.00446/full
[10] Finsterer, J., & Frank, M. (2016). Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therapeutic advances in gastroenterology. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5330602/
[2] National Organization for Rare Disorders. (2017). Rare Disease Data Base: Maple Syrup Urine Disease. Retrieved from https://rarediseases.org/rare-diseases/maple-syrup-urine-disease/#supporting-organizations
[3] Abiri, M., Karamzadeh, R., Mojbafan, M., et al. (August 2016). In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Retrieved from https://link.springer.com/article/10.1007%2Fs11011-016-9867-1
[4] Human Protein Atlas. (n.d.) DBT. Retrieved from https://www.proteinatlas.org/ENSG00000137992-DBT/cell
[5] Evarsson, A., Chuang, J., Wynn, R., et al. (March 2000). Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Retrieved from https://www.sciencedirect.com/science/article/pii/S0969212600001052
[6] Kegg Pathway. https://www.genome.jp/kegg-bin/show_pathway?ko00280+K09699
[7] GRPEL2 GrpE like 2, mitochondrial [ Homo sapiens (human) ]. Retrieved from: https://www.ncbi.nlm.nih.gov/gene
[8] String. https://string-db.org/cgi/network.pl?taskId=IdKnOD6d1YVA
[9] Gilles, Hepple, & T., R. (2016, September 20). Editorial: Mitochondria in Skeletal Muscle Health, Aging and Diseases. Retrieved from https://www.frontiersin.org/articles/10.3389/fphys.2016.00446/full
[10] Finsterer, J., & Frank, M. (2016). Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therapeutic advances in gastroenterology. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5330602/
