This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison. https://genetics564.weebly.com/
What is Phylogenetics?
The general term, phylogeny, is defined as the analysis of species’ relationships. This information is used to study evolutionary patterns. [1] Phylogenetics specifically analyzes sequences of DNA and/or protein to determine the relatedness of species. [2] This information allows for construction of phylogeny trees to visualize the relationship on the feature, gene, protein, etc. of interest. [1]
How do you construct a phylogeny tree?
First, a sequence has to be aligned based on what is the most similar. Single nucleotide/amino acid mutations, insertions, and deletions are taken into account. Sequences can be aligned through computer programs such as ClustalOmega and Mega.
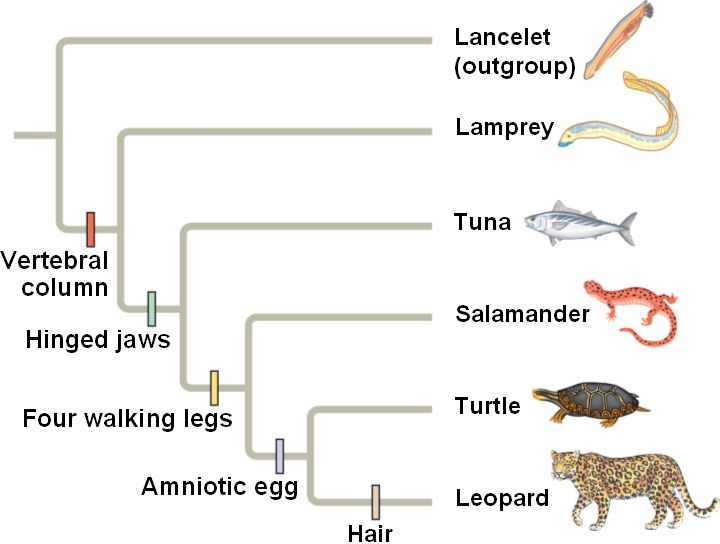
One way to construct a tree is through maximum likelihood. Maximum likelihood is, as the name implies, based on the most likely path. The higher the chance that something diverged at that time, in that order determines the branching of the tree. [3] Computer programs, such as MEGA and ClustalOmega can be used to generate the tree.
Another way to construct a tree is through neighbor joining. Neighbor joining is determined from the differences between two sequences over the total sequence/number of features being analyzed. This is done for every species being compared and a matrix is made. Using the quotient, the tree can be determined based on the smallest change/total. These are joined as “neighbors”; thus, the smaller the distance, the closer the neighbors. [4,5] This again, can be done computationally through the same computer programs as before to construct a tree that best lays out the calculated distance.
Another way to construct a tree is through neighbor joining. Neighbor joining is determined from the differences between two sequences over the total sequence/number of features being analyzed. This is done for every species being compared and a matrix is made. Using the quotient, the tree can be determined based on the smallest change/total. These are joined as “neighbors”; thus, the smaller the distance, the closer the neighbors. [4,5] This again, can be done computationally through the same computer programs as before to construct a tree that best lays out the calculated distance.
Phylogenetic Results of the Human DBT Gene
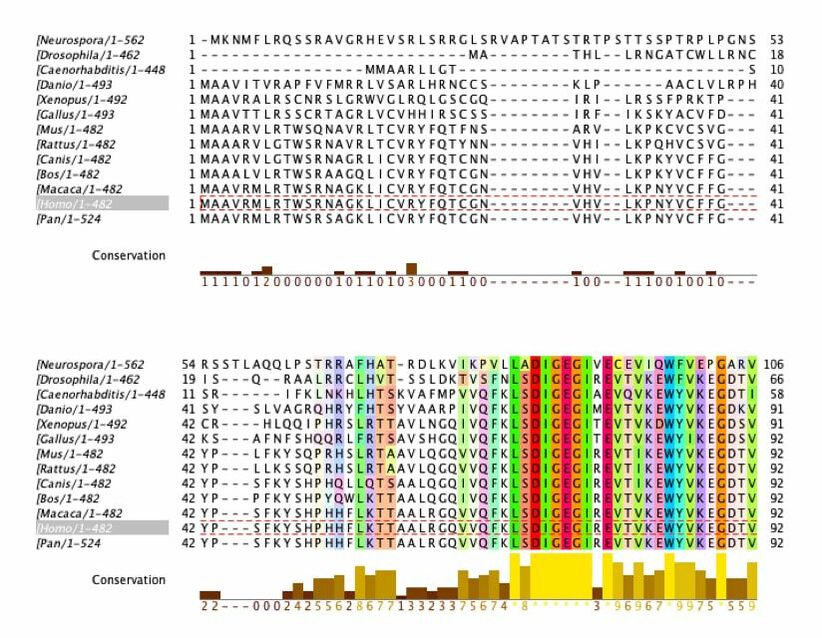
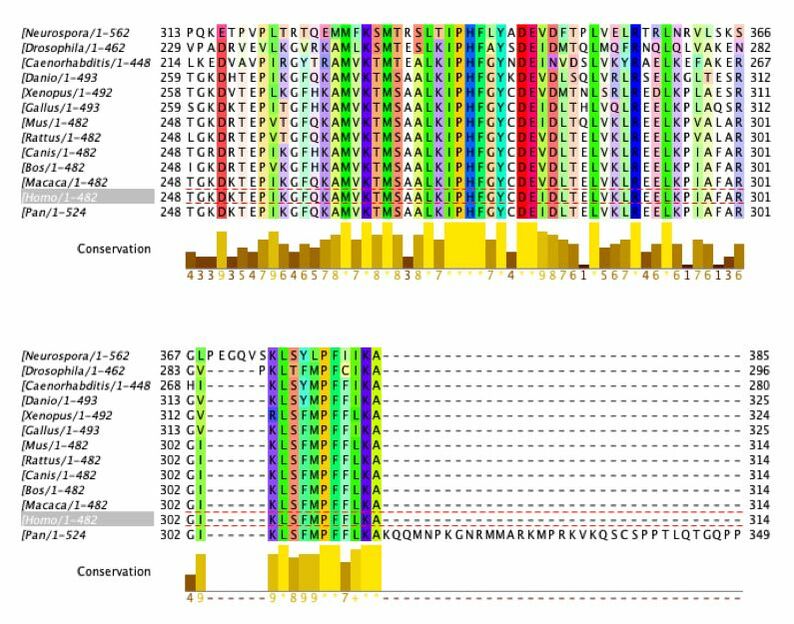
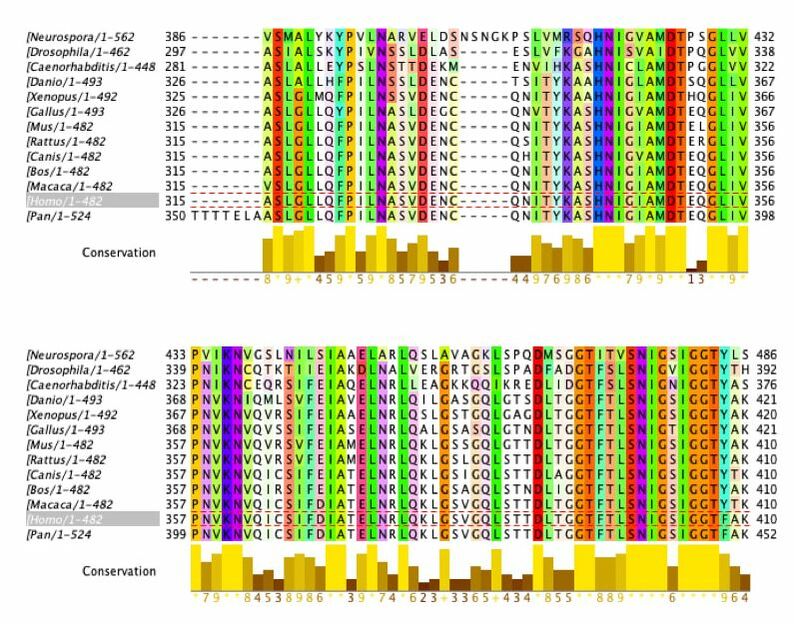
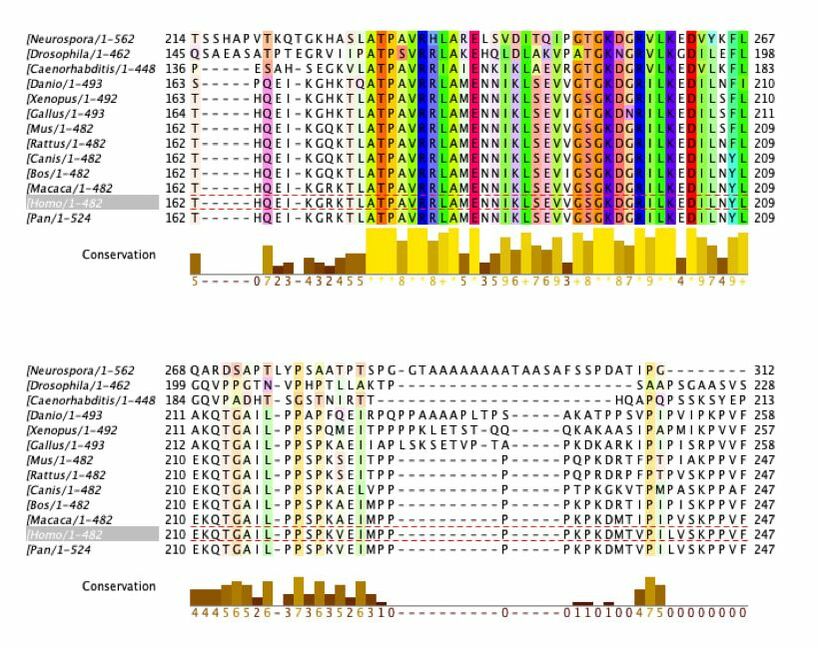
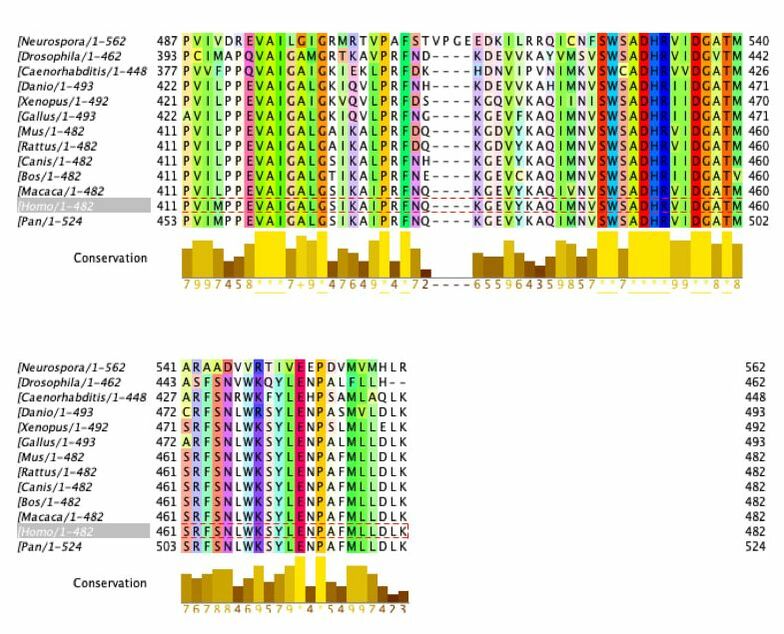
Sequence Alignment with ClustalOmega and Jalview
|
|

|
|
|
|
Phylogenetic Trees Constructed with MEGA
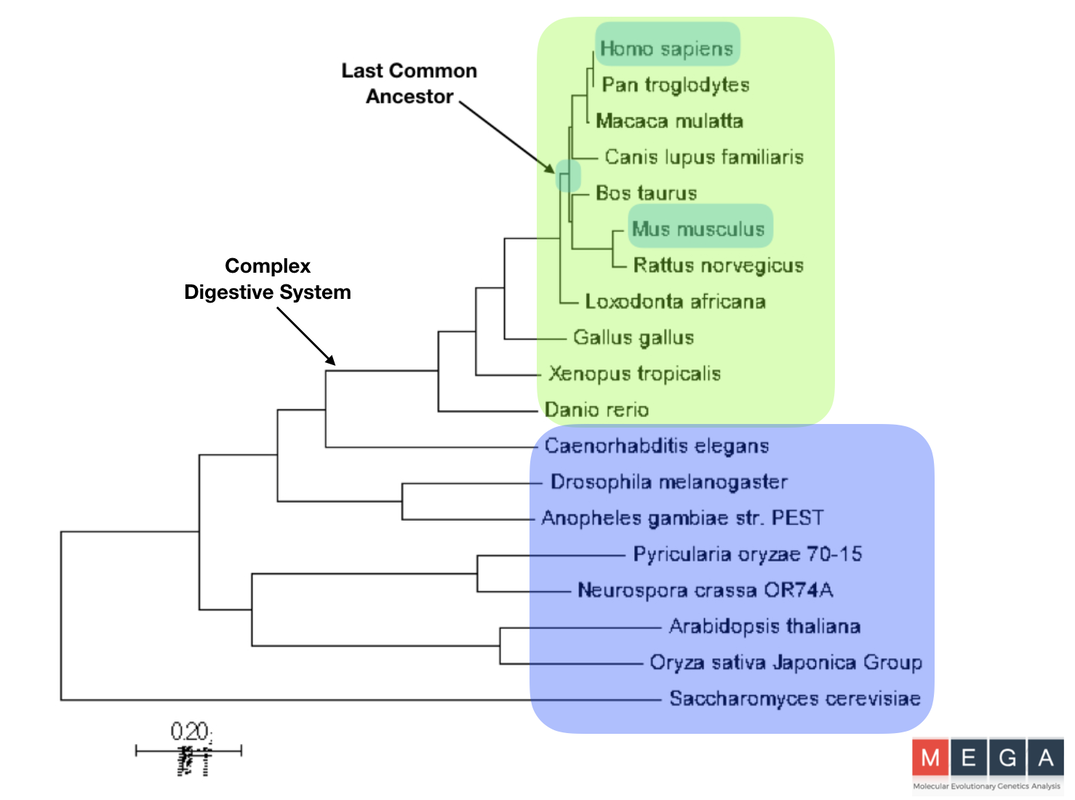
Phylogenic tree constructed using the Maximum Likelihood Method on MEGA software
|
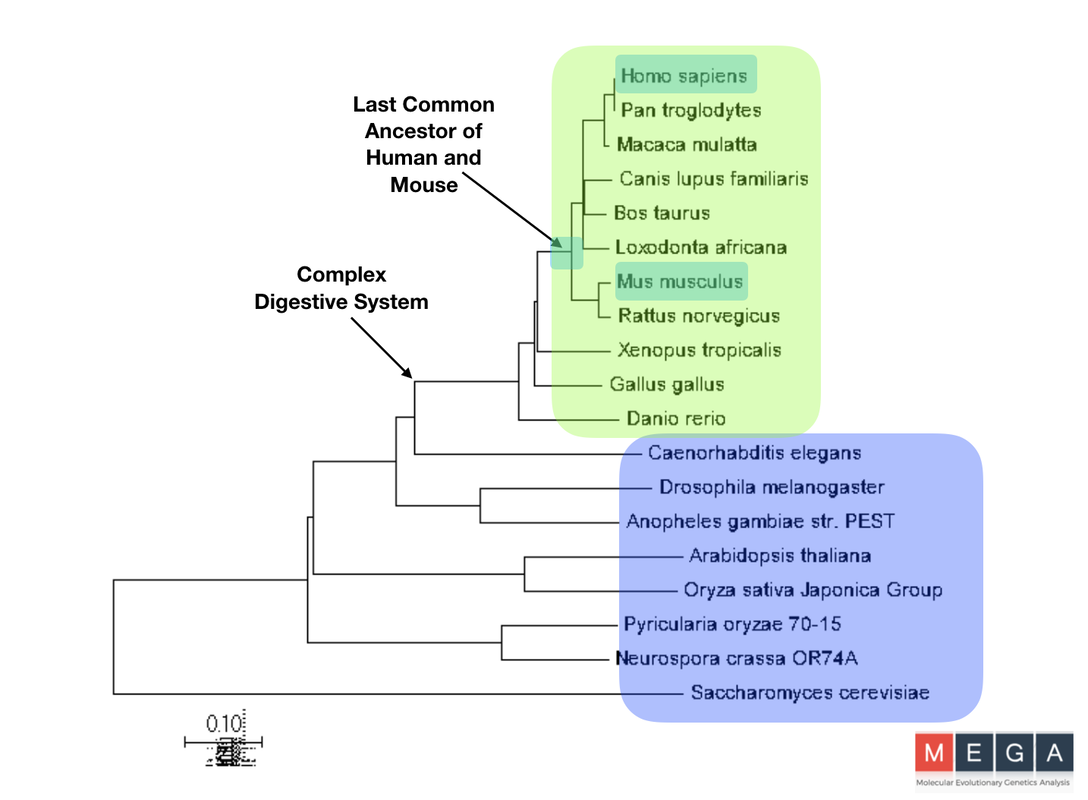
Phylogenic tree constructed using the Neighbor-Joining Method on MEGA software
|
Amino Acids Conserved Among Species with Complex Digestive Systems

Amino acids that are conserved in species with complex digestive systems but not those with simple digestive systems (examples outlined in grey)
Discussion
This data provide important information about conservation of the DBT gene and protein product. The amino acid sequence was analyzed. The ClustalOmega alignment can provide insight into what is highly conserved (yellow graphs underneath sequences). There are large sections of high conservation. These areas have the potential to be classified as domains or be researched to find function. This can be used to provide insight to what areas are conserved among species with complex digestive systems but not the simple digestive systems. As shown above, there are regions where this occurs.
The phylogenic trees visualize how closely related each species’s version of the DBT protein is related. It was found that out of the model organisms listed, the mouse shares the most recent common ancestor with humans in both graphs. This makes sense, as they often are characterized has the model organism having homologous physiological function to humans. This does not show evolutionary tracks of the organisms, only the DBT protein. Using both the Maximum Likelihood and Neighbor-Joining methods, two different paths were found, but are very similar. Although there are slight differences, both trees have a clear divergence of species with complex and simple digestive systems. This conservation and clear divergence is important information to support the idea that there is some part of the DBT gene that plays a role in feeding behavior. If it was not, there would likely not be a clear divergence.
The phylogenic trees visualize how closely related each species’s version of the DBT protein is related. It was found that out of the model organisms listed, the mouse shares the most recent common ancestor with humans in both graphs. This makes sense, as they often are characterized has the model organism having homologous physiological function to humans. This does not show evolutionary tracks of the organisms, only the DBT protein. Using both the Maximum Likelihood and Neighbor-Joining methods, two different paths were found, but are very similar. Although there are slight differences, both trees have a clear divergence of species with complex and simple digestive systems. This conservation and clear divergence is important information to support the idea that there is some part of the DBT gene that plays a role in feeding behavior. If it was not, there would likely not be a clear divergence.
References:
[1] Berkeley University. (n.d.) The family tree. Retrieved from https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_04[2] EMBL-EBI. (n.d.) Phylogenetics: An introduction. Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[3] NCBI. (July 2004). Maximum Likelihood. Retrieved from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
[4] Yang Z, Rannala B. (March 2012). Molecular phylogenetics: principles and practice. Retrieved from https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
[5] RPI.edu. (no date). Phylogentics trees. [Online Slides]. Retrieved from http://www.bioinfo.rpi.edu/bystrc/courses/biol4540/lecture.pdf
Mega Software: https://www.megasoftware.net
ClustalOmega: https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20190308-024243-0917-65706474-p2m&showColors=true&tool=clustalo
Images:
Header: tree-of-life_2000.png
[1] Berkeley University. (n.d.) The family tree. Retrieved from https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_04[2] EMBL-EBI. (n.d.) Phylogenetics: An introduction. Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[3] NCBI. (July 2004). Maximum Likelihood. Retrieved from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
[4] Yang Z, Rannala B. (March 2012). Molecular phylogenetics: principles and practice. Retrieved from https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
[5] RPI.edu. (no date). Phylogentics trees. [Online Slides]. Retrieved from http://www.bioinfo.rpi.edu/bystrc/courses/biol4540/lecture.pdf
Mega Software: https://www.megasoftware.net
ClustalOmega: https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20190308-024243-0917-65706474-p2m&showColors=true&tool=clustalo
Images:
Header: tree-of-life_2000.png

{kind=link}